Vyvíjíme pokročilé (nano)materiály a (nano)technologie, které umožňují řešit klíčové společenské výzvy v oblasti energetiky, chemických technologií a zdravotnictví. Náš výzkum se zaměřuje na poznání a cílené modifikování struktury hmoty na atomární a molekulární úrovni, což nám umožňuje racionálně navrhovat funkční materiály s vlastnostmi přizpůsobenými konkrétním potřebám.
K dosažení těchto cílů využíváme široké a vzájemně se doplňující spektrum teoretických a výpočetních metod, které nám umožňují studovat materiály napříč různými časoprostorovými měřítky:
- Metody výpočtu elektronové struktury: jednorefereční a více-referenční Hartreeho–Fockovy a post-Hartreeho–Fockovy metody
- Teorie funkcionálu hustoty (DFT) a pokročilé metody včetně GW, RPA, BSE
- Semiempirické, empirické a hrubozrnné modely
- Klasické a kvantové simulace molekulární dynamiky
- Výpočty atomistické a spinové dynamiky
- Hybridní výpočty propojující vysoce výkonné (HPC) a kvantové technologie
- Umělá inteligence a metody strojového učení v oblasti návrhu nových materiálů a modelování řízeného daty
Kombinací fyzikálně podložených teoretických poznatků s nejmodernějšími výpočetními technologiemi propojujeme základní porozumění nanotechnologiím s jejich praktickým využitím v reálném světě.
Portfolio
Jednoatomové katalýzy
Navrhujeme a vyvíjíme jednoatomové katalyzátory ukotvené na grafenu s vysokým obsahem defektů a na dalších dvourozměrných (2D) materiálech, čímž dosahujeme mimořádných elektrokatalytických vlastností. Náš výzkum se zaměřuje na klíčové reakce spojené s přeměnou a uchováváním energie, jako jsou vývoj vodíku, redukce kyslíku a redukce oxidu uhličitého, a také na další chemické procesy významné pro chemický a farmaceutický průmysl.
S využitím pokročilých výpočetních metod odkrýváme základní mechanismy, které řídí katalytickou aktivitu. Mapujeme reakční cesty, identifikujeme kroky určující rychlost reakce a odhalujeme, jak lokální koordinace a elektronová struktura izolovaných kovových atomů ovlivňují adsorpci, stabilizaci meziproduktů a reaktivitu v reálných provozních podmínkách.
Naše výpočetní znalosti umožňují řízení interakcí mezi kovem a nosičem i architektury aktivních míst na atomární úrovni. Díky tomu můžeme racionálně navrhovat jednoatomové katalyzátory, které vykazují vysokou aktivitu, mimořádnou selektivitu a dlouhodobou stabilitu pro udržitelné elektrochemické technologie.


Přeměna a uchování energie
Zkoumáme zejména nízkodimenzionální materiály pro elektrochemické uchovávání a přeměnu energie, se zaměřením na superkondenzátory nové generace, baterie a elektrokatalytické energetické technologie.
S využitím teorie funkcionálu hustoty (DFT), molekulární dynamiky a multiškálového modelování navrhujeme materiály s cíleně modifikovanou elektronovou strukturou, povrchovou chemií a vlastnostmi transportu iontů a náboje.
Díky úzké spolupráci s předními experimentálními laboratořemi propojujeme výpočetní screening s nejpokročilejšími metodami syntézy a charakterizace materiálů. To nám umožňuje vyvíjet vysoce výkonné, odolné a udržitelné elektrochemické energetické systémy.
Senzorika
Navrhujeme senzory založené na nanomateriálech, které využívají chemicky funkcionalizované nanostruktury a rozhraní na bázi grafenu k dosažení vysoce selektivního molekulárního rozpoznávání a spolehlivého přenosu signálu.
Pomocí výpočtů elektronové struktury a atomistických simulací odhalujeme základní mechanismy, které řídí povrchovou chemii, termodynamiku adsorpce a procesy v excitovaných stavech. Na těchto základech pak stavíme strategie pro cílenou funkcionalizaci a návrh architektur převodníků, které senzorům propůjčují výjimečnou citlivost a selektivitu.
Hlavním zaměřením naší práce je také modelování fotoluminiscenčních uhlíkových teček a souvisejících uhlíkových nanomateriálů. Objasňujeme, jak složení, povrchové stavy a okolní prostředí ovlivňují optickou emisi. Toto porozumění nám umožňuje cíleně navrhovat sondy pro měření pH, detekci iontů a metabolitů a fluorescenční zobrazování s měřením doby trvání fluorescence v komplexních prostředích.
Díky kombinaci optických a elektrochemických senzorických metod podporujeme vývoj nové generace nanosenzorů pro biomarkery, environmentální kontaminanty a klinicky významné analytické látky. Využíváme k tomu mimořádný měrný povrch, cíleně modifikovatelnou elektronovou strukturu a bohaté fotofyzikální vlastnosti nanomateriálů na bázi uhlíku

Nekovalentní interakce
Ve výzkumu využíváme nejmodernější kvantově-mechanické metody k popisu mezimolekulárních interakcí s výjimečnou přesností. Kombinací teorie spřažených klastrů (např. CCSD(T)) se symetricky adaptovanou poruchovou teorií (SAPT a DFT-SAPT) dokážeme rozložit komplexní interakce na jejich základní složky – elektrostatiku, výměnnou interakci, indukci a disperzi. Tato unikátní schopnost nám umožňuje odhalit fyzikální síly, které určují nekovalentní vazbu. Tyto metody aplikujeme k pochopení vodíkových vazeb, π-stackingu, σ-děrových interakcí a dalších slabých sil, které řídí strukturu, stabilitu a funkci biomakromolekul, nukleových kyselin, protein-ligandových komplexů a pokročilých nanomateriálů.
Na základě tohoto kvantitativního porozumění nekovalentním silám vyvíjíme skórovací funkce založené na kvantové mechanice pro virtuální screening a počítačový návrh léčiv. Tyto přístupy stále častěji rozšiřujeme do oblasti chemie materiálů, kde využíváme nekovalentní interakce na kovových a uhlíkových površích k ladění elektronových a spinových stavů ve funkčních materiálech.
Kvantové výpočty
Náš výzkum se zaměřuje na vývoj a nasazení algoritmů pro kvantové počítače určených k řešení problémů elektronové struktury tam, kde klasické metody přestávají být spolehlivé, zejména u silně korelovaných a multireferenčních reakčních a disociačních drah. S využitím hardwarově optimalizovaných kvantových obvodových ansatzů, jako je unitární Cluster–Jastrowova vlnová funkce, a zpřesňujících strategií založených na algoritmu NOQE (Non-Orthogonal Quantum Eigensolver), počítáme profily potenciální energie základních a excitovaných stavů malých molekul (např. NO a CO2), které slouží jako referenční benchmarky. To zahrnuje i modelování jevů, jako je vyhnuté křížení, větvení reakčních kanálů, a švů v ohnutém stavu, které určují reaktivitu. Tyto algoritmy doplňujeme o praktické pracovní postupy pro kompilaci a zmírňování chyb – optimalizované pro hardware na bázi iontových pastí s plnou konektivitou.
Naším cílem je poskytovat experimentálně ověřené hodnoty energií, definovat robustní metriky přesnosti a stanovit protokoly pro odhad výpočetních zdrojů. Tyto protokoly pak umožňují přechod k výpočetně náročnějším modelům s větším aktivním prostorem a ke kvantovým zařízením příští generace.


Magnetismus 3D a 2D materiálů
Zkoumáme magnetické jevy ve 3D i 2D materiálech, van der Waalsových heterostrukturách a komplexních intermetalických systémech. Při výzkumu kombinujeme rozsáhlé výpočty v rámci relativistické teorie funkcionálu hustoty (DFT) s metodami výpočtu elektronové struktury přesahující rámec DFT.
Náš výzkum se zaměřuje na klíčové mechanismy ovládající magnetismus, včetně magnetické anizotropie, výměnných interakcí, vazby mezi spinem a mřížkou a souhry mezi lokalizovanými a itinerantními magnetickými momenty. Studujeme širokou škálu materiálů významných pro spintroniku – od vrstevnatých magnetických materiálů přes systémy se vzácnými zeminami a 4f a 5f elektrony až po vysokoentropické slitiny.
Propojením dynamiky mřížky z prvních principů, magnetické stabilizace zprostředkované fonony a magnetoelastických a magnetokalorických jevů se spinovými a spin-mřížkovými simulacemi při konečných teplotách odhalujeme ultrarychlou a teplem řízenou dynamiku magnetizace. Tyto poznatky slouží jako vodítko pro návrh materiálů se stabilním magnetickým uspořádáním pro účely magnetického chlazení a ukládání dat.
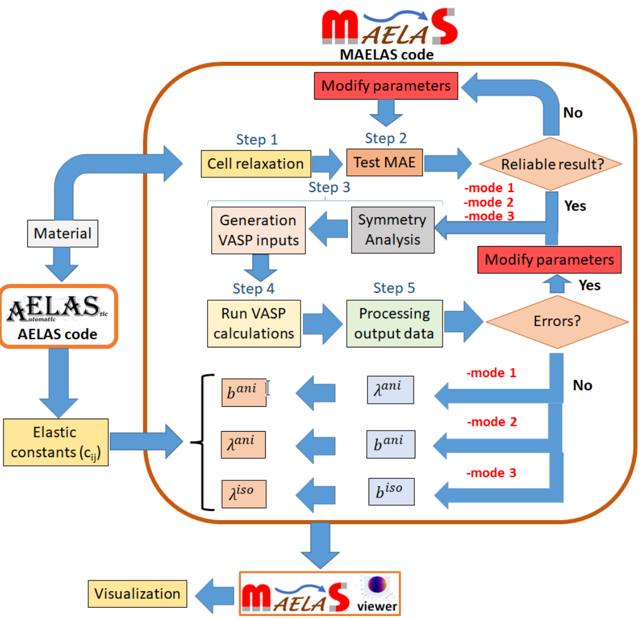
Magnetostrikce a magnetoelasticita
Zkoumáme vzájemné působení magnetismu a elasticity (tzv. magnon-fononové jevy), které umožňuje přímou přeměnu mechanické energie na magnetickou a naopak (převodníky). Toho se využívá v široké škále aplikací, včetně rotačních a lineárních motorů, senzorů síly, energeticky úsporných elektronických zařízení, aktuátorů, magnetoakustických zařízení (sonary), seismických detektorů a dalších technologií.
Náš výzkum se zaměřuje na klíčové mechanismy ovládající magnetokrystalovou anizotropii a její závislost na deformaci, jak se projevuje v rámci magnetoelastických jevů. To zahrnuje studium izotropních a anizotropních koeficientů magnetostrikce a jejich původu v elektronové struktuře, stejně jako vývoj nových metod, které přibližují výpočty reálným podmínkám.
Využitím atomistických výpočtů, spinové dynamiky a metod strojového učení v kombinaci s DFT výpočty elektronové struktury vyvíjíme spin-mřížkové modely pro feromagnetické i antiferomagnetické materiály. Tyto modely umožňují provádět rozsáhlé simulace vybraných magnetických materiálů zahrnující miliony atomů, a to přímo na úrovni inženýrského návrhu.
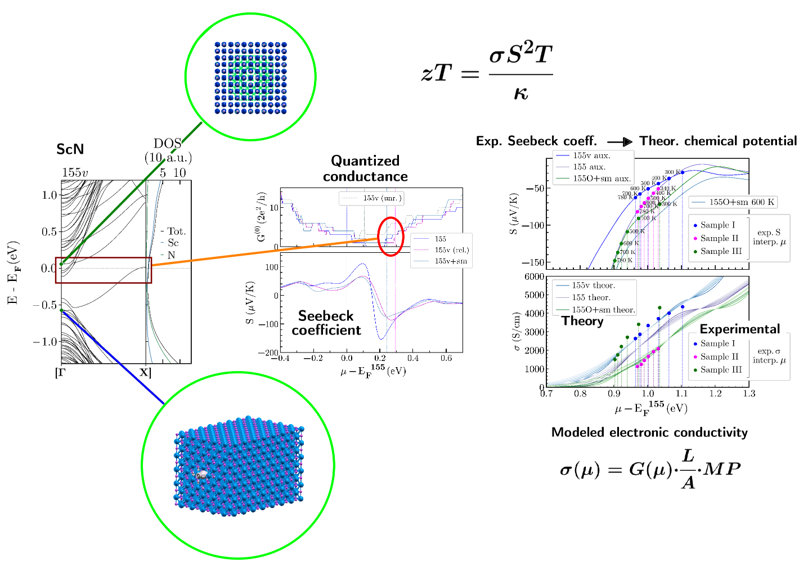
Termoelektřina
Termoelektřina představuje přímou přeměnu tepla na elektřinu. Vysoká termoelektrická účinnost (zT) vyžaduje kombinaci dobré elektrické vodivosti (σ) a nízké tepelné vodivosti (κ). Nitridy přechodných kovů jsou slibnou třídou materiálů pro aplikace při středních až vysokých teplotách, protože mohou vykazovat příznivý Seebeckův koeficient (S) spolu s vysokou elektrickou vodivostí (σ).
Hlavní výzvou zůstává skutečnost, že přenos tepla v těchto materiálech je stále příliš vysoký pro dosažení efektivní termoelektrické účinnosti. Možným řešením je využití metod přípravy (tenkých vrstev), které umožňují snížit přenos tepla, aniž by došlo k narušení elektrického přenosu. Klíčovou roli v tomto procesu hrají defekty a nečistoty. Zkoumáme vzájemné působení těchto faktorů a jejich dopad na termoelektrickou účinnost v nově vyvíjených materiálech na bázi nitridových tenkých vrstev.