We develop advanced (nano)materials and (nano)technologies to address key societal challenges in energy, chemical technologies, and healthcare. Our research focuses on understanding and controlling matter at the atomic and molecular level, enabling the rational design of functional materials with tailored properties.
To achieve these goals, we employ a broad and complementary set of theoretical and computational methods, spanning multiple length and time scales:
- Electronic structure methods: single- and multireference Hartree–Fock and post-Hartree–Fock approaches
- Density Functional Theory and beyond including also GW, RPA, BSE methods
- Semiempirical, empirical, and coarse-grained interaction models
- Classical and quantum molecular dynamics simulations
- Atomistic and Spins dynamics calculations
- High Performance / Quantum hybrid computing
- Artificial intelligence and machine-learning techniques for materials discovery and data-driven modeling
By combining physically grounded theory with state-of-the-art computational approaches, we bridge fundamental understanding and real-world applications of nanotechnologies.
Portfolio
Single atom catalysis
We design and engineer single-atom catalysts anchored on defect-rich graphene and two-dimensional materials to unlock exceptional electrocatalytic performance. Our research targets key energy-relevant reactions, including hydrogen evolution, oxygen reduction, and carbon dioxide reduction, as well as other chemical reactions relevant for chemical and pharmaceutical industry.
Using advanced computation methods, we uncover the fundamental mechanisms that control catalytic activity. We resolve reaction pathways, identify rate-determining steps, and reveal how the local coordination and electronic structure of isolated metal atoms govern adsorption, intermediate stabilization, and reactivity under realistic operating conditions.
Our computational insights enable atomic-level control of metal–support interactions and active-site architectures, guiding the rational design of single-atom catalysts that deliver high activity, outstanding selectivity, and long-term durability for sustainable electrochemical technologies.


Energy Conversion and Storage
We explore namely low-dimensional materials for electrochemical energy storage and conversion, targeting next-generation supercapacitors, batteries, and electrocatalytic energy technologies.
Using density functional theory, molecular dynamics, and multiscale modeling, we design materials with tailored electronic structure, surface chemistry, and ion and charge transport properties. Our simulations provide deep mechanistic insight into processes at electrified interfaces, linking atomic-scale phenomena to device-level performance.
Through close collaboration with leading experimental laboratories, we connect computational screening with state-of-the-art materials synthesis and characterization, enabling the development of high-performance, durable, and sustainable electrochemical energy systems.
Sensing
We design nanomaterial-based sensors that exploit chemically functionalized nanostructures and graphene-derived interfaces to achieve highly selective molecular recognition and robust signal transduction.
Using electronic structure calculations and atomistic simulations, we uncover the fundamental mechanisms governing surface chemistry, adsorption thermodynamics, and excited-state processes. These insights guide the rational design of functionalization strategies and transducer architectures, enabling enhanced sensitivity and specificity.
A major focus of our work is the modeling of photoluminescent carbon dots and related carbon-based nanomaterials, where we resolve how composition, surface states, and local environment control optical emission. This understanding enables the targeted design of probes for pH sensing, ion and metabolite detection, and fluorescence-lifetime imaging in complex media.
By integrating theoretical insights across optical and electrochemical sensing modalities, we support the development of next-generation nanosensors for biomarkers, environmental contaminants, and clinically relevant analytes, leveraging the exceptional surface area, tunable electronic structure, and rich photophysics of carbon-based nanomaterials.

Noncovalent Interactions
Our research employs state-of-the-art quantum-mechanical methods to describe intermolecular interactions with exceptional accuracy. By combining coupled-cluster theory (e.g., CCSD(T)) with symmetry-adapted perturbation theory (SAPT and DFT-SAPT), we can decompose complex interactions into their fundamental components – electrostatics, exchange, induction, and dispersion. This unique capability allows us to reveal the physical forces that govern noncovalent binding. We apply these methods to understand hydrogen bonding, π-stacking, σ-hole interactions, and other weak forces that control the structure, stability, and function of biomacromolecules, nucleic acids, protein–ligand complexes, and advanced nanomaterials.
Building on this quantitative understanding of noncovalent forces, we develop quantum-mechanics-based scoring functions for virtual screening and computational drug design. These approaches are increasingly extended to materials chemistry, where noncovalent interactions at metallic and carbon-based surfaces are exploited to tune electronic and spin states in functional materials.
Quantum computing
Our research develops and deploys quantum-computing algorithms for electronic-structure problems where classical methods become unreliable, especially along strongly correlated and multireference reaction and dissociation pathways. Using hardware-efficient circuit ansätze such as the unitary Cluster–Jastrow wavefunction and refinement strategies based on the Non-Orthogonal Quantum Eigensolver, we compute ground- and excited-state potential-energy profiles for small molecules that serve as stringent benchmarks (e.g., NO and CO₂), including avoided crossings, branching channels, and bent-state seams that control reactivity. We pair these algorithms with practical compilation and error-mitigation workflows—optimized for trapped-ion hardware with all-to-all connectivity – to deliver experimentally validated energies, define robust accuracy metrics, and establish resource-estimation protocols that guide scaling to larger active spaces and next-generation quantum devices.


Magnetism of 3D and 2D materials
We explore magnetic phenomena in 3D as well as in 2D materials, van der Waals heterostructures, and complex intermetallic systems, combining large-scale relativistic density functional theory with beyond-DFT electronic-structure methods.
Our research targets key mechanisms governing magnetism, including magnetic anisotropy, exchange interactions, spin–lattice coupling, and the interplay between localized and itinerant magnetic moments. We study a broad range of materials relevant to spintronics, from layered magnetic materials and rare-earth and 4f/5f-electron systems to high-entropy alloys.
By integrating first-principles lattice dynamics, phonon-mediated magnetic stabilization, and magnetoelastic and magnetocaloric effects with finite-temperature spin and spin–lattice simulations, we uncover ultrafast and thermally driven magnetization dynamics. These insights guide the design of materials with robust magnetic order in order to provide magnetic cooling and data information storage.
Magnetostriction and Magnetoelasticity
We explore the interplay between magnetism and elasticity (magnon-phonon) phenomena, where the interplay between mechanical and magnetic properties affects each other. It allows for direct conversion between the mechanical and magnetic energy (transducers). This is exploited in the range of applications including rotational and linear motors, force sensors, low-energy demanding electronics, actuators, magneto-acoustic devices (sonars), seismic detectors, etc.
Our research aims at key mechanisms governing magnetocrystalline anisotropy, its strain dependence as exhibited by the magnetoelastic phenomena, including isotropic and anisotropic magnetostriction coefficients and their electronic structure origin as well as developing novel approaches that brings computing closer to the real-world conditions.
By leveraging the atomistic and spin-dynamics calculations and ML techniques of the DFT electronic structure calculations we develop spin-lattice models for ferromagnetic as well as antiferromagnetic materials that could be used in large-scale computations containing millions of atoms of desired magnetic materials at the engineering design level.
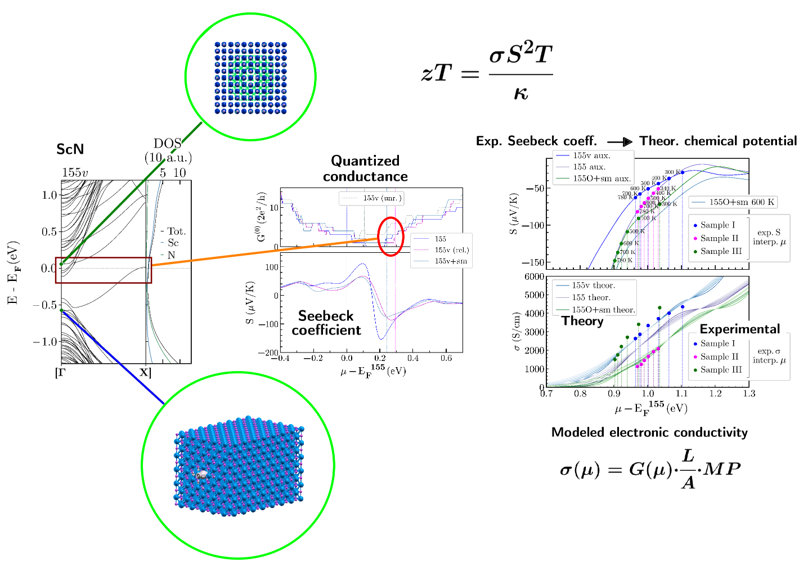
Thermoelectrics
Thermoelectricity is the direct conversion between heat and electricity. High thermoelectric performance (zT) requires a combination of good electrical conduction (σ) and poor heat conduction (κ). Transition-metal nitrides are a promising class of materials for medium- to high-temperature applications, as they can exhibit a good Seebeck coefficient (S) together with high electrical conductivity (σ).
The main challenge is that thermal transport in these materials is still too high for efficient thermoelectric performance. A possible solution is to use preparation techniques (thin-films), that reduce thermal transport without interfering with electrical transport. Defects and impurities play a crucial role in this process. We investigate the interplay between defects and impurities and their impact on thermoelectric efficiency in novel nitride thin-film materials.