Since the advances in HPC the simulation of material behavior has played a key role in our lives. This fact was even more pronounced once there was a way to perform quantum mechanical calculations to obtain electronic structure of materials and its behavior to link to many physical and chemical properties. First-principles (ab initio) calculations at present are the parameter free approach for i) verification of the experiments, ii) to simulate conditions or calculate material properties that are not directly accessible or measurable, iii) to design novel materials. Within the flagship we address fundamental and state-of-the-art topics like to design nuclear fuels materials from radioactive compounds for IVth generation nuclear reactors, ultrafast magnetization dynamics for novel data storage, complex spintronic devices exploiting multiferroicity, and engineering applicable materials at finite temperatures and pressures, e.g. novel permanent magnets.
Researcher:
- Dr Dominik Legut
SCIENTIFIC DIRECTION: LATTICE AND ATOMISTIC VIBRATIONS - 3D AND 2D MATERIALS
Solids, molecules and atoms vibrate even at T=0K around their equilibrium positions. These collective lattice motions (phonons) are responsible for many interesting phenomena like thermal expansion, superconductivity, etc. Due to the computational power of IT4Innovations HPC, these phenomena can be described at an atomistic scale, and thermal expansion and heat capacity of any materials can be determined directly from quantum-mechanical calculations without any empirical parameter. The research team models IR and Raman absorption spectra and mutual interaction of phonons to understand how heat is transported, and what it affects in matter. These calculations allow the design of novel nuclear fuels, searching for ultra-hard solids similar to diamond, as well as improving the figure of merit of thermoelectric materials. Another specific area of interest of our colleagues within this flagship is the study of phonon-electron and phonon-spin interaction. This allows superconductivity to be modeled. Moreover, for the very first time magnetic materials can be modeled under finite temperatures directly using quantum-mechanical calculations aiming to describe the behaviour of magnetocalorics (cooling with a magnetic field, and understanding ultrafast demagnetization by laser pulse for novel data-storage techniques.
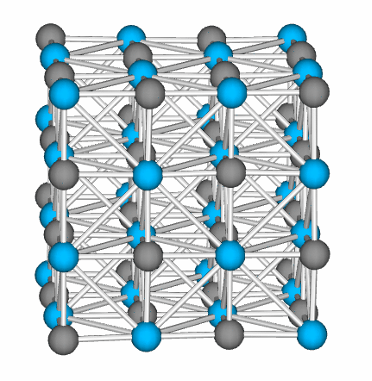
SCIENTIFIC DIRECTION: MAGNETISM STUDIES
The flagship research team also studies, models, and designs magnetic materials at different spatial and time scales for several technological applications such as magnetic recording, spintronics, electric motors, electric generators, magnetic actuators, biomedicine (magnetic hyperthermia), magnetic refrigeration, etc. They apply and combine modeling techniques to describe and understand magnetic materials accurately. Some of these approaches use Density Functional Theory (DFT) to calculate magnetic intrinsic properties at the microscopic scale and novel structures predicted by evolutionary algorithms, atomistic spin dynamics (to take into account finite-temperature effects), molecular dynamics (to study grain boundary phenomena and crystal phase stability), micromagnetics (to calculate magnetic domains, microstructure effects, and hysteresis loops at the macroscopic scale) and multiphysics finite-element models (to simulate magnet performance at operating conditions).
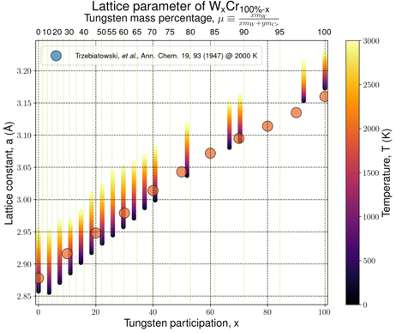
SCIENTIFIC DIRECTION: TRANSITION METAL AND HIGH ENTROPY ALLOYS MODELING
Within the research of clean energy technologies, new alloys for novel thermonuclear fusion reactors, such as ITER (International Thermonuclear Experimental Reactor) are designed using realistic, quantum-mechanical models for real simulation of First Wall alloys in reactor vessels, which are exposed to the heat of hydrogen plasma with a temperature of cca 200,000,000 °C, protected only by an external magnetic field (5T) keeping the plasma confined. Combining Quantum Mechanics, Statistical Physics, and High-Performance Computing, the used methods thus create a kind of a synergic modeling engine. Recently, surprising properties were obtained for 5-components equiatomic alloys stabilized by the entropy term at high temperatures with properties much different to those single element constituents. A new category of so called high (HEA), middle, and low entropy alloys has been instigated. Despite energies mostly being focused on the mechanical properties, magnetic behaviour cannot be omitted, as it is able to influence the phase stability and mechanical properties. The very first HEA is composed of Cr-Mn-FeCo-Ni being surprisingly paramagnetic at RT. Thus the objective is to determine the effect of the composition and substitutions on the magnetic character here.
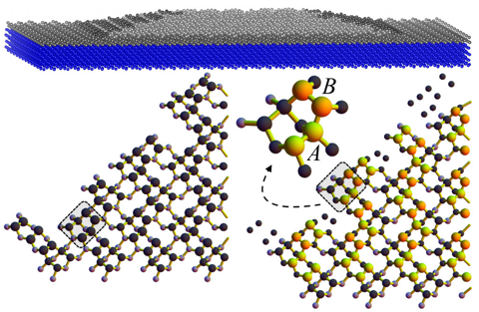
SCIENTIFIC DIRECTION: LARGE-SCALE ATOMISTIC SIMULATIONS
Design and prediction of physical properties for realistic materials requires accurate calculation of large systems of atoms. Electronic structure calculations based, for example, on DFT became, in fact, the main computing method in materials science. However, standard DFT calculations deal with relatively small system sizes of a few hundred atoms, both for the purpose of computing time and memory requirements as computational intensity increases with the square and cube of the number of atoms. For more realistic systems with dislocations, interfaces, grain boundaries, random and diluted alloys, nano-particles, and biomolecules, however, it is necessa - ry to perform simulations of an order of several thousand to several million atoms. The CONQUEST code, utilizing the density matrix approach with a local basis, is a very efficient parallel method that provides a linear scaling solution. Such calculations are capable of an accurate description of the growth processes of nano-sized systems such as nano-particles, quantum dots, and nano-wires.
Another large-scale atomistic modeling method is the molecular dynamics approach based on interatomic potentials. For many applications, it allows physical properties behaviour to be determined under a given temperature (diffusion, melting tempera - ture, coexistence of different phases). In such cases, DFT calculations would again be prohibitively expensive, or even unfeasible due to running long simulations in the order of nanoseconds, even when using the CONQUEST code. Within the research, machine and deep learning methods are applied to design accurate interatomic potentials from DFT calculations.
SCIENTIFIC DIRECTION: CODE DEVELOPMENT
The flagship researchers develop several codes written in the Fortran, Python, and Matlab/Octave programming languages. Below there is a selection of those that have already been published and made publicly available.
- PNADIS: This code is an automatic analyser of the Peierls-Nabarro stress and dislocation cores developed in MATLAB. This code calculates the dislocation core structure, Peierls stress, the pressure field around the dislocation core, and the solid solution strengthening of a crystal based on the Peierls-Nabarro model and other models derived therefrom.
- MAELASviewer: An online application for visualization and analysis of magnetostriction via a user-friendly interactive graphical interface.
MATERIAL DESIGN FOR INDUSTRY: COOPERATION WITH CONTINENTAL CZECH REPUBLIC
To design many technological applications, it is necessary to understand the physical and chemical processes occurring at the macroscopic level. Many miniature electronic devices are embedded into protective materials (silica, polymers, etc.), which may degrade in certain environments (gasoeus, liquid) and under certain conditions (high/low temperature and/or pressure). Such processes can be simulated for realistic models (large systems of 104–106 atoms) using classical Molecular Dynamics (MD) with reactive force field (ReaxFF). This interaction potential is a trade-off between accuracy, provided by DFT calculations, and the system size and the length of the temporal evolution of the system. These simulations allow various interfaces between materials and the environment (gas-solid and/or liquid-solid interface), structural stability under various pressure and temperature conditions, and the diffusion of various molecules into the material and its effects on its properties to be modeled. This knowledge may provide prompt technological solutions for industry (design of new materials, protective layers, and optimal operation conditions) similarly to those designed for the Continental Czech Republic company in 2019.